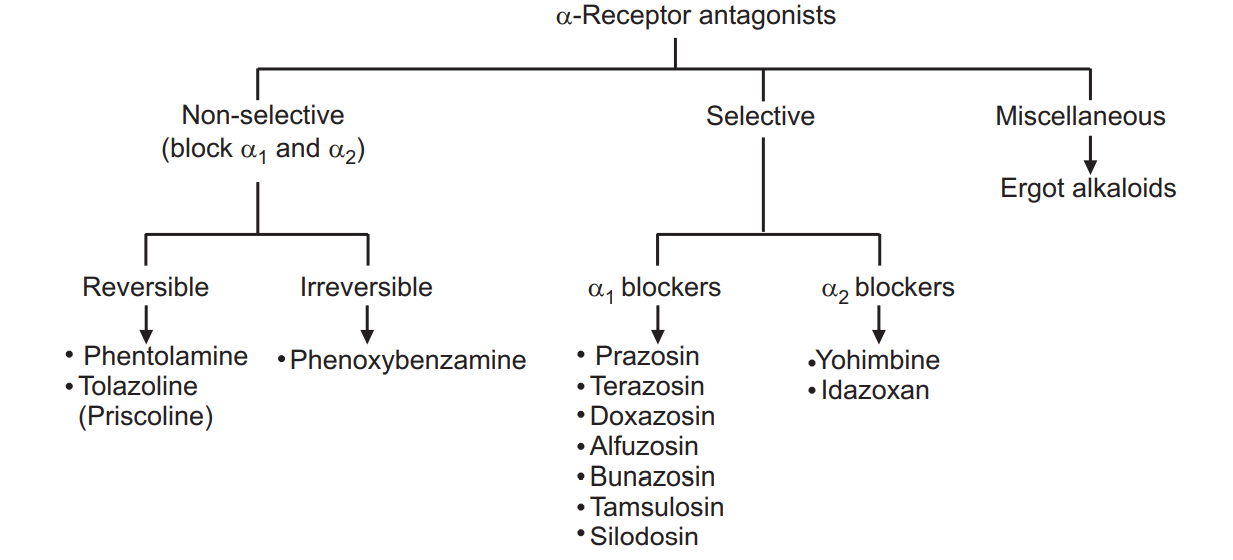
On the basis of their selectivity and mode of action, α-adrenoceptor antagonists can be classified as mentioned below. See Fig. 1.1. Alpha Adrenergic Blockers are classified as.
Table of Contents
(i) Reversible non-selective α-blockers
These drugs are competitive antagonists at α-adrenergic receptors and have similar affinities for α1- and α2-receptors. Two drugs viz. Phentolamine and Tolazoline deserve discussion in this category.
(a) Phentolamine
Cardiovascular effects
Blockade of vasoconstrictor α1-receptors in the periphery by Phentolamine leads to vasodilatation, a decrease in peripheral vascular resistance leading to hypotension. The resultant fall in BP stimulates the baroreceptor reflex causing sympathetic discharge. Since α1-receptors are blocked, the sympathetic discharge stimulates β1-receptors in the heart producing tachycardia. Being non-selective, it also blocks pre-synaptic α2-receptors limiting the neuronal release of NE to produce more tachycardia and palpitation. The patients may suffer from events of postural hypotension. In absence of efficient peripheral vasoconstriction in the erect posture, there is a peripheral pooling of blood leading to cerebral hypoxia, vertigo, and fainting, which are important features of postural hypotension.
Other effects
The following effects are observed with Phentolamine:
- Nasal stuffiness results due to vasodilatation and congestion of nasal mucosa.
- Myosis is due to loss of tone of radial muscles of the iris and unopposed contraction of circular muscle.
- Improved urine flow rates due to relaxation of smooth muscles of the urinary bladder neck and prostate.
- Failure of ejaculation and impotence due to inhibition of contractions of vas deferens and ejaculatory ducts.
- Nausea, vomiting, and diarrhea due to partial inhibition of relaxant sympathetic influences on GIT, and increase in gastric secretions due to agonistic action on histamine H2 receptors.
Pharmacokinetics
It is poorly absorbed from GIT. It is administered intravenously. It has an immediate onset and a shorter duration of action.
Therapeutic uses
- For diagnosis and management of Phaeochromocytoma.
- For peripheral vascular disorders: It is used to treat Raynaud’s syndrome and frostbite.
- To prevent dermal necrosis: It is used subcutaneously to prevent dermal necrosis after incidental extravasation of NE from the intravenous infusion.
- To prevent hypertensive crises following abrupt withdrawal of Clonidine and those resulting from the ingestion of tyramine-containing food with MAO inhibitors. It is available as a 10 mg/ml injection: FENTANOR, FENTOSOL.
(b) Tolazoline
It is similar to Phentolamine, but it is less potent. It is better absorbed from GIT. It is rarely used.
(ii) Irreversible non-selective α-blockers
The only drug in this category is Phenoxybenzamine.
(a) Phenoxybenzamine
It binds covalently to α1- and α2-receptors causing irreversible blockade of the receptors with a longer duration of action (20-48 hours). It inhibits the reuptake of released NE by adrenergic nerve terminals. It also crosses the blood-brain barrier. It causes vasodilatation, a progressive decrease in peripheral resistance, and tachycardia. Cardiovascular effects of an overdose of Phenoxybenzamine cannot be reversed by catecholamines due to covalent binding with the receptors, making it the drug a choice for Phaeochromocytoma. It causes marked postural hypotension due to impairment of compensatory vasoconstrictor responses. Tolerance to it develops later. Other toxic effects include reversible inhibition of ejaculation, salt and water retention, cardiac arrhythmia, sedation, fatigue, and nausea.
Therapeutic uses
- Treatment of Phaeochromocytoma in an intravenous dose of 1 mg/kg, controls severe hypertension during surgery for Phaeochromocytoma.
- To treat a peripheral vascular disease like Raynaud’s syndrome and frostbite. It is used in an oral dose of 10 mg three times a day.
It is available as a 10 mg capsule or 50 mg/ml injection: FENOXENE.
(iii) Reversible, selective α1-blockers
The primary drug in this category is Prazosin. A few derivatives of Prazosin are also available.
(a) Prazosin
It is a selective α1-receptor antagonist. It causes peripheral vasodilatation and a fall in arterial pressure with lesser tachycardia probably because of a lack of α2-receptor blocking actions, limiting the release of NE. It also decreases cardiac preload and suppresses sympathetic outflow from CNS.
It is a potent inhibitor of the enzyme cyclic phosphodiesterase leading to an increase in cAMP. It also causes a rise in the concentration of HDL and a decrease in LDL and triglycerides. Being an α1-selective antagonist, it relaxes smooth muscles of the urinary bladder neck, prostate capsule, and prostatic urethra leading to improvement of urine flow in cases of benign prostatic hypertrophy.
Pharmacokinetics
It is well absorbed after oral administration. Its plasma half-life is about 4 hours.
Therapeutic uses
- Treatment of hypertension: Titration of the dose is needed to limit postural hypotension. Initially, a dose of 1 mg may be given orally at bedtime.
- In the treatment of benign prostatic hyperplagia (BPH). The oral dose of 1-5 mg twice daily is useful.
- In patients with Raynaud’s disease, calcium channel blockers are preferred over Prazosin.
Adverse effects
- The major adverse effect is postural hypotension (Syncopal attack).
- Impotence, nasal congestion, GIT upset, and sodium and water retention are additional adverse reactions.
It is available as a tablet of 1 mg, 2 mg, or 5 mg: MINIPRESS, PRAZOPRESS.
(b) Terazosin and Doxazosin
Both are α1-blockers like Prazosin but have a longer duration of action. Both are well absorbed after oral administration. The plasma half-life of Tetrazosin is 12 hours, and that of Doxasosin is 20 hours. One daily dose is preferred for the treatment of hypertension and BPH. Terazosin in a dose of 2-5 mg or Doxazosin in a dose of 1-4 mg, once daily is useful to improve urine flow. It may be combined with another drug Finasteride, which inhibits the conversion of testosterone to dihydrotestosterone. The combination is useful in reducing the progress of BPH.
Terazosin: HYTRIN, TERAPRESS, TERALFA, OLYSTER is available as the tablet of 1 mg, 2 mg, and 5 mg; Doxasosin: DOXACARD, DURACARD is available as the tablet of 1 mg, 2 mg, and 4 mg
(c) Bunazosin and Alfuzosin
Both are orally effective α1-blockers similar to Prazosin. Alfuzosin is given at a dose of 2.5 mg three times a day. It has a shorter half-life of 4 hours and hence needs frequent administration. It should be avoided in patients with hepatic impairment, due to extensive metabolism by the liver. Bunazosin has a slightly longer half-life. Both drugs are used to treat BPH.
Alfuzosin: ALFOO, FUAL, AFFUSIN, and PROFUZO is available as a 10 mg extended-release tablet.
(d) Tamsulosin and Silodosin
These are selective α1-blockers. Tamsulosin is more effective for BPH than for hypertension. It has a plasma half-life of 8 hours and better bioavailability than Prazosin. Abnormal ejaculation and intra-operative floppy iris syndrome (which causes problems during cataract surgery) are major adverse reactions. Silodosin is a weaker but long-acting analog of Tamsulosin.
Tamsulosin: CONTIFLO-OD, URIPRO, VELTAM, URIMAX is available as 0.2 mg, 0.4 mg tablet.
(iv) α2-receptor blockers
The only drug in this category is Yohimbine.
Yohimbine
It is a natural alkaloid available from Pausinystalia Yohimbe. It is lipid soluble. It crosses the blood-brain barrier(BBB). It has antagonistic action to 5-HT. It can be used to treat autonomic insufficiency by promoting NE release by blocking pre-synaptic α2-receptor. It can also be used to treat male sexual dysfunction and to treat diabetic neuropathy and postural hypotension. It can abruptly reverse the anti-hypertensive effect of clonidine, which is a notable drug interaction. Its clinical role needs to be established.
(v) Miscellaneous non-selective α-blockers
Ergot alkaloids fall in this category.
Ergot alkaloids
Alkaloids from ergot like Ergotamine and Dihydroergotamine exhibit complex pharmacological actions. They block both α1– and α2-receptors. In addition, they act as partial agonists to α-receptors and 5-HT2 receptors. Some of them are Oxytocics and dopamine, receptor agonists. Methysergide, a synthetic compound is related to ergot alkaloids. It is a potent 5-HT antagonist and was used for migraine prophylaxis. Ergonovine/Ergometrine is an ergot alkaloid. Its derivatives methyl-ergonovine and dihydroergotamine are used for their uterine relaxant (tocolytic) action.
Make sure you also check our other amazing Article on: Classification of Neurotransmitters