
Evaluation of drugs ensures the identity of a drug and determines the quality and purity of drugs. The main reasons behind the need for evaluation of crude drugs are biochemical variation in the drug, effect of treatment and storage of drugs, and the adulterations and substitutions.
- Organoleptic Evaluation
- Microscopical Evaluation
- Chemical Evaluation
- Physical Evaluation
- Biological Evaluation
Table of Contents
Organoleptic And Morphologic Evaluation
The organoleptic character (also known as sensory evaluation) and morphological character (also known as macroscopic method) are the preliminary identification methods. These two methods (organoleptic and morphologic methods) are together known as diagnostic characters of the crude drugs. These refer to the evaluation of drugs by systemic morphological characters such as leaves, barks, fruits, flowers, roots, and rhizomes, etc. (by size, shape, and special features) and further identification by sensory characters like color, odor, taste, and consistency (touch and texture). The general appearance of the crude drug indicates it is likely to comply with the standard. For example, fractured surfaces in cinchona and cascara are important characteristics. Seeds of different species of caraway, dill, leaves of senna species, the disc-shaped structure of nuxvomica, the conical shape of aconite, etc., can be distinguished. Leaves of different species of menthe can be differentiated by smell. A smell can also identify the adulterated crude drugs like clove and exhausted clove. By taste, bitter drugs like quassia, chirata, kalmegh, neem, sweet tasted drugs like glycyrrhiza, Stevia, pungent tasted drugs like ginger, capsicum, spice drugs like black pepper, nut meg, caraway, etc. can be identified. Texture and nature of fracture also provide important information like liquorice is hard and its fracture is fibrous. Fracture of ipecac is brittle, aconite and nuxvomica are horny, etc.
Microscopic Evaluation
This evaluation is also known as anatomical evaluation or histological evaluation of crude drug This method can be used to identify the organized drug in powdered form by their histological characters or anatomical cell or tissue arrangements. Various reagents (like chloral hydrate, conc. HCl, glycerin) and stains (phloroglucinol for identification of lignified cells and tissues like xylem, phloem, etc., Iodine for detection of starch grains, etc.) can be used to differentiate cellular structure. This evaluation also covers the study of the constituents by application of the chemical method to small quantities of the powdered drug. This is known as chemomicroscopy. The elements such as stomata, trichomes, vessels, fibers, stone cells, starch grains, medullary rays, oil cells, calcium oxalate crystals are present in powdered conditions and are used in the microscopic identification of crude drugs. For example, leaves contain epidermis cells including trichome, stomata, calcium oxalate crystals (if present); bark contains phloem elements (cinchona), stone cells (Kurchi) or sometimes both (cascara); trichome (nuxvomica); fruits contain oil cells, endosperm, pericarp, epicarp, mesocarp, etc. This evaluation method is also useful in the identification of closely related drugs or species. Hence microscopy is two types viz. Qualitative and Quantitative. Qualitative microscopy is only detection and identification of cellular structures of drugs whereas Quantitative microscopy is to determine particular cellular substances like linear measurement as the diameter of starch grains, length of fibers, vessels, quantitative microscopic constant as the stomatal index, vein islet number, palisade ratio, Lycopodium spore method, etc. using camera lucida method using two types of micrometers viz. Stage and eyepiece micrometers (Figs. 1.1, 1.2, and 1.3).
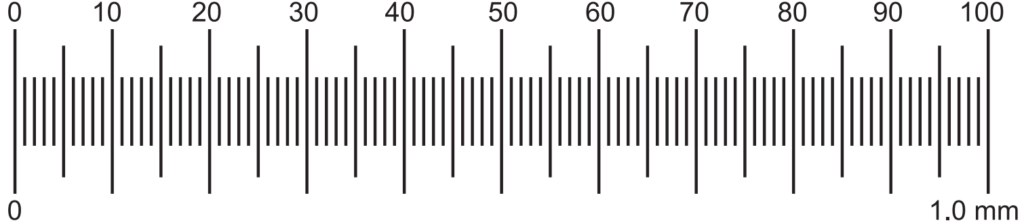
Each big division of stage micrometer is a total of 1.0 mm in length. It has 100 divisions. Each division is 1/100 = 0.01 mm.
Eyepiece micrometer: It is a circle of glass with a scale etched on the surface. It is a scale of 1 mm in length with 100 divisions.
Calibration
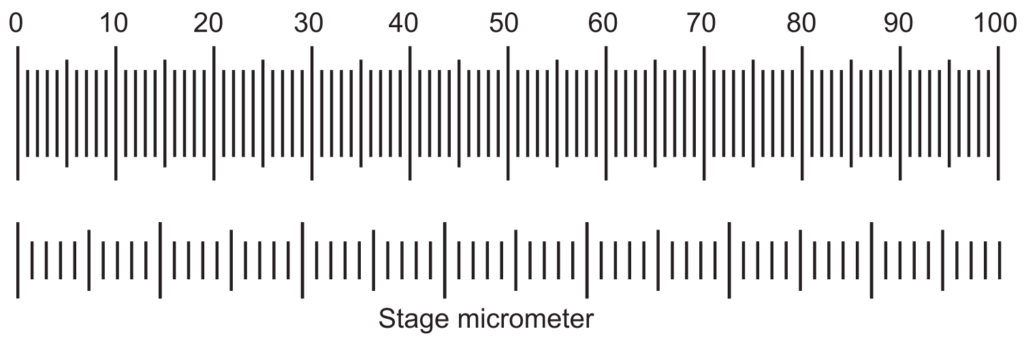
Both the eyepiece and stage micrometers are kept in their respective position and coincided with any line of the eyepiece and any line of stage micrometer. Thereafter the number of lines counted which coincides further (Fig. 1.3). Then the divisions of the eyepiece and those of stage micrometer present between the two coincided lines are counted.
Now, if 7 divisions of eyepiece coincided with 4 divisions of stage micrometer,
Then the value of one division of the eyepiece micrometer is calculated as:
1 division of stage micrometer = 0.01 mm = 10 µ
Therefore, 4 division of stage = 10 × 4 = 40 µ
Now, 7 division of eyepiece = 4 division of stage = 40 µ Hence, 1 division of eyepiece = 40/7 = 5.71 µ
This is known as the calibration factor.
With the help of this calibration factor (5.71 µ) further quantitative microscopy like starch grain diameter, fiber length determination, etc. are carried out. For these two experiments, there is no required camera lucida. Directly by observed sample slide calculated the length or diameter of the sample with the help of eyepiece micrometer and readings are recorded. Finally, all the readings are multiplied by the calibration factor.
Example: Starch grain diameter of one starch is measured by eyepiece micrometer are 8 divisions. Then 8 × 5.71 µ = 45.68 µ is the actual dimension of one starch grain.
Camera Lucida
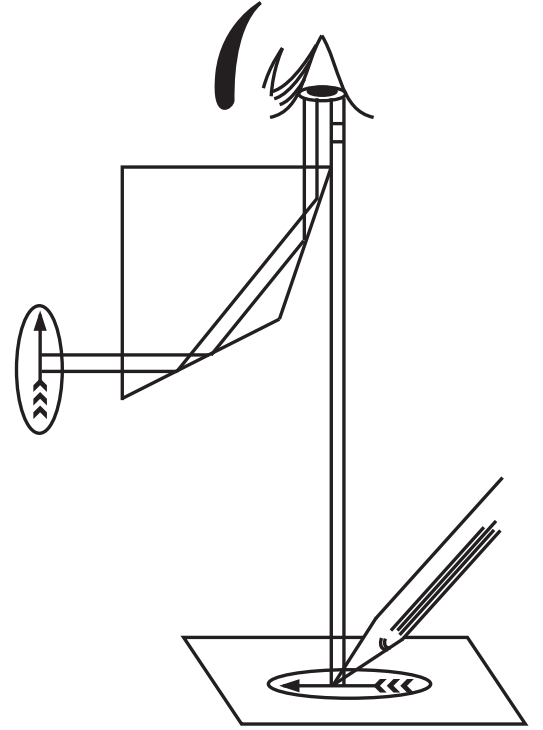
It is an optical device or instrument in which rays of light are reflected by a prism to produce an image on a sheet of paper, from which a drawing is made. It works on a simple optical principle reflecting the beam of light through a prism and a plane mirror.
There are two types of camera lucida namely Swift Ives and Abbe model camera lucida. The Abbe camera lucida consists of a prism fitted over the eyepiece of the microscope. A sidearm is carrying a mirror that is supported vertically over the tracing paper (Fig. 1.4 b and c).

In Swift Ives Camera lucida the plane mirror is replaced by a small right-angled prism. It is a small size and fits over the eyepiece of the microscope with a screw.
Principle of Camera lucida
During use, the light from the drawing board is reflected by the plane mirror into the prism and further reflected into the observer’s eye that is seeing the drawing paper and the pencil in the direction of the stage of the microscope. The prism has a small opening through which the observer is seeing the image of the object. As a result, the superimposed image then conveniently traces the microscopic object (Fig. 1.5).
Stomata
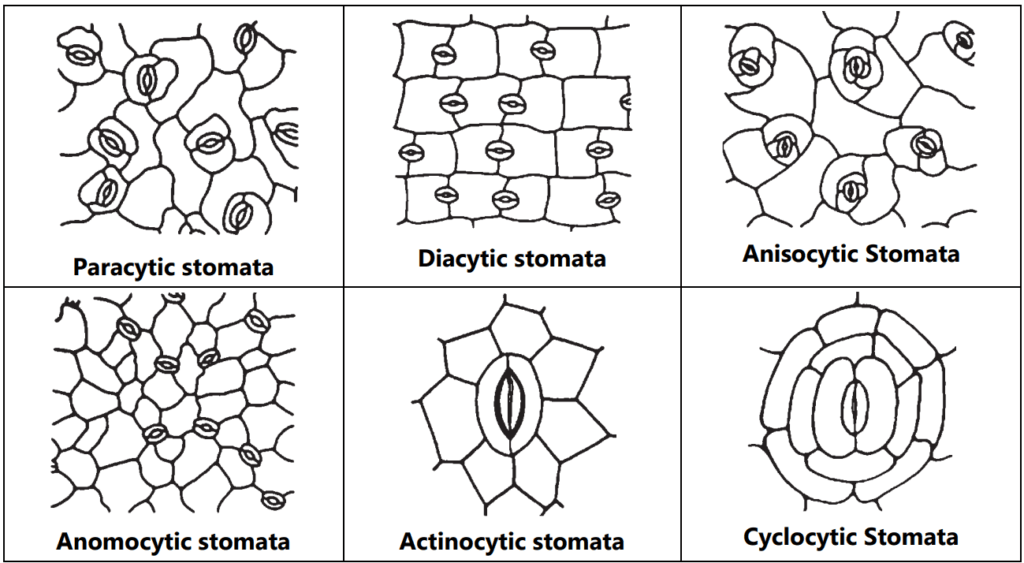
Stomata are the minute epidermal opening present in the aerial part of the leaves. Mainly it helps in gaseous exchange. It consists of two kidney-shaped cells with a middle tiny pore. Broadly four types of stomata viz. Moss type, Gymnospermous type, Gramineous type, and Dicotyledonous type. As per the arrangement of stomata in dicot plants, they are classified into paracytic (stomata two guard cells covered by two subsidiary cells, e.g.: Senna), Diacytic (Guard cell is covered by two subsidiary cells on the right angle, e.g.: Peppermint), Anisocytic (Stomata number of guard cells are two but covered by three subsidiary cells, e.g.: Datura), Anomocytic (Stomata is surrounded by varying number of subsidiary cells, e.g.: Digitalis) and Actinocytic (Two guard cells are surrounded by radiating subsidiary cells, e.g.: Banksia conferta), Cyclocytic (stomata is surrounded by four or more subsidiary cells, e.g.: Baccharis articulata) (Fig. 1.6).
Leaf constants
Stomal Index:
The stomatal index is the percentage of the number of stomata formed by the total number of epidermal cells, including the stomata, each stoma being counted as one cell. Place leaf fragments of about 5 × 5 mm in size in a test tube containing about 5 mL of chloral hydrate solution and heat in a boiling water bath for about 15 minutes or until the fragments become transparent. Transfer a fragment to a microscopic slide and prepare the mount, the lower epidermis uppermost, in chloral hydrate solution and put a small drop of glycerol-ethanol solution on one side of the cover-glass to prevent the preparation from drying.

Where S = the number of stomata in a given area of the leaf; and E = the number of epidermal cells (including trichomes) in the same area of the leaf.
Stomatal number:
It can be defined as the average number of stomata per square millimeter area of epidermis cell. The stomatal number can be determined by treating the lamina of the leaves with chloral hydrate and then the part of the epidermal surface is removed carefully and observed under the microscope.
Place leaf fragments of about 5 × 5 mm in size in a test tube containing about 5 mL of chloral hydrate solution and heat in a boiling water bath for about 15 minutes or until the fragments become transparent. Transfer fragments to a microscopic slide and prepare the mount the lower epidermis uppermost, in chloral hydrate solution and put a small drop of glycerol-ethanol solution on one side of the cover glass to prevent the preparation from drying. Examine with a 40x objective and a 6x eyepiece, to which a microscopical drawing apparatus is attached. Mark on the drawing paper a cross (x) for each stomata, and calculate the average number of stomata per square millimeter for each surface of the leaf.
Palisade ratio:
The palisade ratio is the average number of palisade cells under one epidermal cell. Place leaf fragments of about 5 × 5 mm in size in a test tube containing about 5 mL of chloral hydrate solution and heat in a boiling water bath for about 15 minutes or until the fragments become transparent. Transfer a fragment to a microscopic slide and prepare the amount of the upper epidermis in chloral hydrate solution and put a small drop of glycerol solution on one side of the cover glass to prevent the preparation from drying. Examine with a 40x objective and a 6x eyepiece, to which a microscopical drawing apparatus is attached. Trace four adjacent epidermal cells on paper; focus gently downward to bring the palisade into view and trace sufficient palisade cells to cover the area of the outlines of the four epidermal cells. Count the palisade cells under the four epidermal cells. Where a cell is intersected, include it in the count only when more than half of it is within the area of the epidermal cells. Calculate the average number of palisade cells beneath one epidermal cell, dividing the count by 4; this is the “Palisade ratio”. For each sample of the leaf make no fewer than ten determinations and calculate the average number.
Examples: Atropa belladonna: 6 – 10; Datura stramonium: 4 – 6; Mentha piperita: 5.1 – 8.1.
Vein Islet Number:
The mesophyll of a leaf is divided into small portions of photosynthetic tissue by a stomosis of the veins and veinlets; such small portions or areas are termed “Vein-Islets”. Vein-islets present in per square millimeter is known as “Vein-Islet number”. This value has been shown to be constant for any given species and, for full-grown leaves, to be unaffected by the age of the plant or the size of the leaves. The vein-islet number has proved useful for the critical distinction of certain nearly related species.
Take pieces of leaf lamina with an area of not less than 4 square millimeters from the central portion of the lamina, excluding the midrib and the margin of the leaf. Clear the pieces of lamina by heating in a test tube containing chloral hydrate solution on a boiling water bath for 30 to 60 minutes or until clear and prepare amount in glycerol-solution or, if desired, stain with safranin solution and prepare the mount in Canada balsam. Place the stage micrometer on the microscope stage and examine with 4x objective and a 6x eyepiece. Draw a line representing 2 mm on a sheet of paper by means of a microscopical drawing apparatus and construct a square on the line representing an area of 4 square millimeters. Move the paper so that the square is seen in the center of the field of the eyepiece. Place the slide with the cleared leaf piece on the microscope stage and draw in the veins and veinlets included within the square, completing the outlines of those vein-islets which overlap two adjacent sides of the square. Count the number of vein-islets within the square including those overlapping on two adjacent sides and excluding those intersected by the other two sides. The result obtained is the number of vein-islets in 4 square millimeters. For each sample of leaf, make no fewer than three determinations and calculate the average number of vein-islets per square millimeter.
Examples: Cassia angustifolia: 19 – 23; Digitalis purpurea: 2 – 5.5; Cassia acutifolia: 25-30.
Lycopodium spore method:
This method is used to identify the crude drugs when the chemical and physical methods are inapplicable. This method is also useful to detect the adulteration present in the crude drugs containing starch grains.
Examples: Adulterated drug-containing starch can be determined by counting the number of starch grains per mg and calculating the amount from the known number of starch grains per mg of the pure starch. The percentage purity of an authentic powdered ginger is calculated using the following equation:
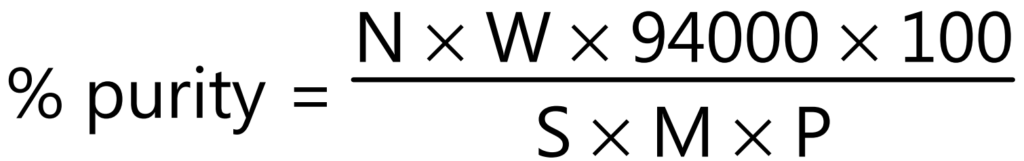
Where,
N = Number of characteristic structures (starch grain) in 25 fields.
W = Weight in mg of lycopodium taken.
S = Number of lycopodium spores in the same 25 fields.
M = Weight in mg of the sample, calculated on the basis of sample dried at 105°C.
P = 2,86,000 in case of ginger starch grains powder.
Significance:
- Determination of foreign organic matter.
- Determination of percentage purity of drugs.
- Detection of adulterant
Physical Evaluation
Physical standards are to be determined for drugs that are constant for crude drugs and help in drug evaluation. Physical constants such as specific gravity, refractive index, swelling factor, optical rotation, viscosity, etc., are useful in determining the identity, purity, and quality of the drugs (Figs. 1.7 and 1.8).
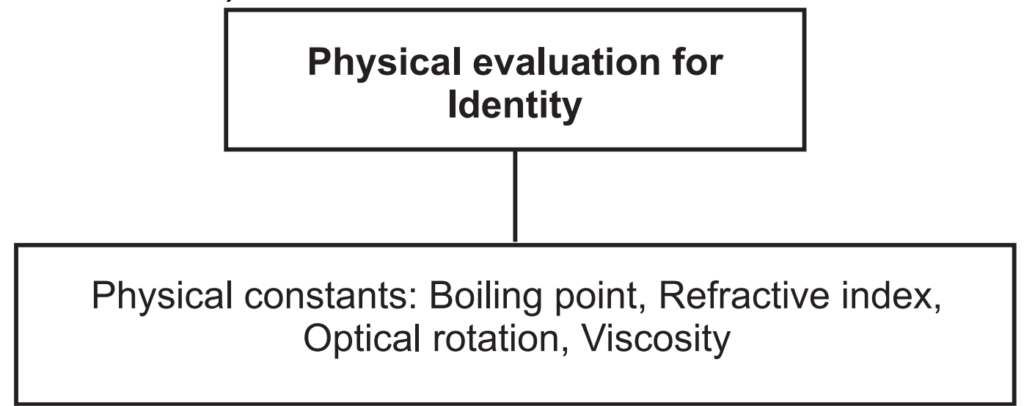
Boiling point: The boiling point of a substance is the temperature at which the vapor pressure of the liquid equals the pressure surrounding the liquid and the liquid changes into a vapor. A liquid in a vacuum has a lower boiling point than when that liquid is at atmospheric pressure. Boiling points are approximately related to their molecular weight. The higher the molecular weight, the higher is the boiling point.
Refractive index (Ri): This method is used for the determination of refractive index to essential and fixed oils and also for detection of adulteration present in the oils. It is defined as the ratio between the velocity of light in the air and in the oil. It can be determined with a refractometer. It is denoted as, n = sin i/sin r. Where, n = refractive index, sin i = sine of the angle of incidence, and sin r = sine of the angle of refraction. It is measured at 20°C ± 5°C temperature. Sometimes Abbe refractometer is used for accurate measurements of Ri.
Examples: Clove oil: 1.530 – 1.531 at 20°C., Castor oil: 1.475 – 1.479 at 20°C.
Optical rotation: Basically volatile oils show the ability to rotate in the plane of polarized light to the left or right side, and likewise, they are called laevorotatory or dextrorotatory respectively. It is measured by using a polarimeter at 20°C.
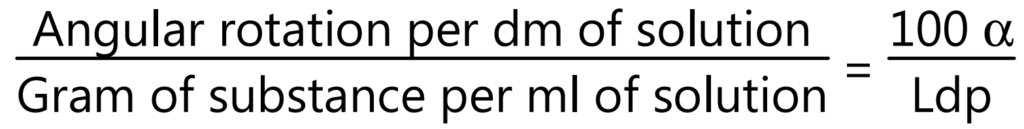
where, α = Observed rotation, L = Length of the observed layer in drug, d = Density, and p = gm of substance in 100 g of solution.
Viscosity: Viscosity is a measure of the resistance of a fluid which is being deformed by either shear or tensile stress. The most common method of determining kinematic viscosity in the lab utilizes the capillary tube viscometer.
Examples: Clove oil: 0° to – 1.5°, Cinnamon oil: 0° to – 2°.
Melting Point: The melting point of a solid is the temperature at which it changes state from solid to liquid. At the melting point, the solid and liquid phases exist in equilibrium. The melting point of a substance depends (usually slightly) on pressure and is usually specified at standard pressure. The freezing point or crystallization point is the temperature of the reverse change from liquid to solid. It can be determined by the capillary method. In this method, a thin glass capillary tube containing a compact column of the substance to be determined is introduced into a heated stand (liquid bath or metal block) in close proximity to a high accuracy thermometer. The temperature should continue at a fixed rate until the sample in the tube transitions into the liquid state. While determining the melting point, several observations and the temperatures are recorded.
Examples: Cocoa butter: 30 – 35°C; Wool fat: 34 – 44°C.
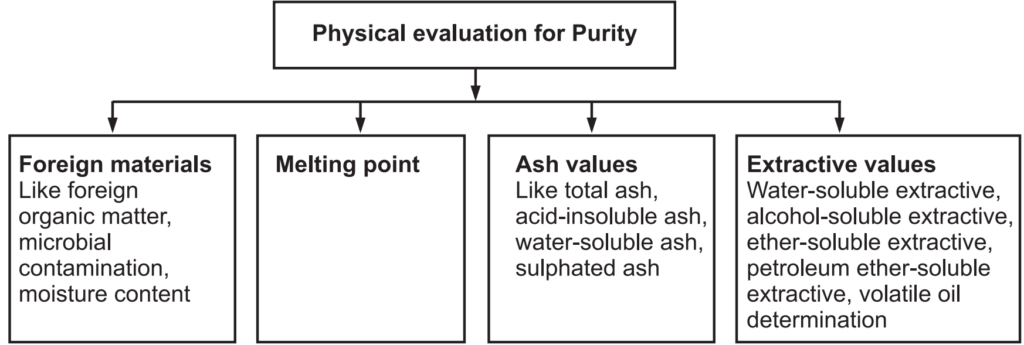
Foreign Organic Matters: Anything extra present in the drug which does not comply with the authentic drug may be considered as a foreign matter. The foreign matter can be present in the drug due to improper harvesting. The source of foreign organic matter can be animal excreta, insect, or mold and is determined by sedimentation or floatation method.
Examples: Curcumin – not more than 2.0%; Neem – not more than 1.5%.
Ash Value: After the incineration of the crude drugs, the remnants are known as ash. Ash contains mostly inorganic salts. It gives the idea about the quality and purity of crude drugs. It can be determined by (a) Total ash: Carbon and organic matters of the crude drugs are converted to ash at a temperature of about 900°C. It mostly contains carbohydrates, phosphates, silicates, and silica. This total ash is further used with water-soluble ash and acid-insoluble ash. Water-soluble ash is produced by separating the water-soluble materials, which is dried to yield water-soluble ash. Further, total ash, when treated with dilute hydrochloric acid, removes many inorganic salts to yield mainly silica in the residue. This is known as acid-insoluble ash. When the crude drug is incinerated with dilute sulphuric acid at a temperature above 600°C, the ash obtained by this method is known as sulfated ash. This method converts all oxides and carbonates to sulfate salt.
Total Ash Value: 2 g of the ground air-dried sample is to weigh into previously ignited, dried, and tarred silica crucible. Spread the material in an even thin layer. It is then kept on a gas burner under a low flame and is ignited slowly to obtain a carbonized residue. It is then placed in the muffle furnace and the temperature of the muffle is adjusted to 450- 500°C and ignition is then allowed in the muffle furnace. After 3 hours, switch off the muffle furnace and allow it to cool, and remove the silica crucible. Further, cool in a desiccator and weigh. If carbon-free ash cannot be obtained in this manner, moist residue with 2 mL of water or saturated solution of ammonium nitrate. (This is to expose bits of unashed carbon). Then it is dried on a water bath and then on the hot plate, and after that ignite in a muffle furnace to constant weight. Remove from muffle furnace and allow cooling for 30 minutes in a desiccator and weight. Finally, the percentage of ash is calculated.
Acid Insoluble Ash: To the silica crucible containing the total ash obtained, 25 mL of hydrochloric acid (~70 g/l) is added. Cover with a watch glass and boil gently for 5 minutes on a hot plate or burner. Rinse the watch glass with 5 mL of hot water and add these washings to the crucible. The insoluble matter will collect on an ash-less filter paper by filtration and rinse the filter paper repeatedly with hot water until the filtrate becomes neutral/free from acid. Transfer the filter paper containing the insoluble matter to the original crucible, dry on a hot plate, and ignite to a constant weight in the muffle furnace at 450- 500ºC. Remove the silica crucible from the muffle furnace and allow cooling the matter in a desiccator for 30 minutes, and then weighing without delay. The content of acid-insoluble ash will be calculated as a percentage.
Water-Soluble Ash: The ash is boiled for a few minutes with the required amount of water, collect the insoluble matter in a silica crucible and then wash with hot water and ignite for 15 minutes at a temperature not exceeding 450°C. The weight of the insoluble matter will be subtracted from the weight of the ash. The difference in weight represents the water-soluble ash. The percentage of water-soluble ash is calculated with reference to the air-dried drug.
Calculation:
Total ash/Acid insoluble ash/Water-soluble ash % = (B − C) × 100/A
Where, A – Sample weight in g; B – Weight of dish + contents after drying (g) C – Weight of empty dish (g).
Determination of Sulphated Ash:
Heat a silica or platinum crucible to redness for 10 minutes, allow to cool in a desiccator, and weigh. Put 1 to 2 g of the substance, accurately weighed, into the crucible; ignite gently at first, until the substance is thoroughly charred. Cool, moisten the residue with a few mL of sulphuric acid and then ignite at 800º ± 25ºC until all black particles have disappeared. Conduct the ignition in a place protected from air currents. Allow the crucible to cool, add a few drops of sulphuric acid, and heat. Ignite as before, allowing cooling and weighing. Repeat the operation until two successive weighings do not differ by more than 0.5 mg. Examples: Ashwagandha.
Significances of Ash Values:
- Determine authenticity and purity of crude drug sample
- Determine the presence of inorganic salts like carbonates, phosphates, silicates, etc. in crude drugs.
- Useful to detect adulterated products.
- Useful to detect exhausted products.
- Useful to detect earthy matter in the products.
Moisture Content: Moisture content in the drug provides an enzymatic activity or facilitates the growth of microbes, which results in deterioration. The crude drugs were heated at 105°C to constant weight and calculated the total loss of weight. The methods used for the determination of moisture contents are (a) Loss on drying (b) the Azeotropic distillation method (c) Karl Fischer method (d) Directly with the moisture pan balance. The Toluene distillation method is used for the determination of moisture content in the volatile oil-containing drugs.
2 gm of the powdered drug is transferred into a shallow weighing bottle and the contents will be distributed evenly to a depth not exceeding 10 mm. The full bottle is then heated at 105°C in a hot air oven and weighed until a constant weight is obtained. The difference is calculated from the weight after drying and the initial weight is the moisture content. The amount of calculated moisture content is confirmed with moisture pan balance.
Ultrasound wave for moisture content: air-coupled ultrasonic spectroscopy is enabled ultrasonic waves to be applied to the on-line and real-time assessment of the water content of different fresh plant materials. Examples: Senna leaf: Not more than 10% w/w; Digitalis: not more than 5% w/w; Aloes: not more than 10% w/w.
Significances of Moisture Content:
- Excess of moisture present in drugs is leads to degradation of quality.
- Excess moisture content is leads to a microbial attack on the drugs
- Moisture content determination is also helpful in the quality of the drug.
Extractive Values: All the chemical constituents are soluble either in polar, semi-polar, or organic solvents. Total soluble constituents of the drug in any particular solvent or mixture of solvents may be called its extractive value or percent extractive.
Alcohol Soluble Extractive Value:
Prepare coarse powder of air-dried drug. Take 100 mL of ethanol (specified strength) in a conical flask. Macerate 5 g of powdered drug in a conical flask, close the flask for 24 hours. Shake the flask frequently during the first 6 hours; allow it to stand for 18 hours. Filter rapidly taking precaution against loss of ethanol. Evaporate 25 mL of the filtrate to dryness in a tarred flat bottomed shallow dish. Dry at 105°C and weigh it. Calculate the % of alcohol-soluble extractives with reference to the air-dried drug.
Water Soluble Extractive Value:
Prepare coarse powder of air-dried drug. Take 100 mL of chloroform water in the conical flask. Macerate 5 g of powdered drug in a conical flask, and close the flask for 24 hours. In between the flask is shaken for the first 6 hours and then stands for 18 hours. Filter rapidly by decanting the water extract and then evaporating 25 mL of the filtrate to dryness in a tarred flat bottomed shallow dish. Dry at 105°C and weigh it. Calculate the % of water-soluble extractives with reference to the air-dried drug.
Significances of Extractive Values:
- This method is important when the constituents of drugs can’t be readily estimated by any other means.
- It indicates the nature of chemical constituents present in drugs.
- It helps in the identification of adulterants.
Chemical Evaluation
With this method active constituents in the drugs are determined. There are various methods for chemical evaluation (Fig. 1.9).
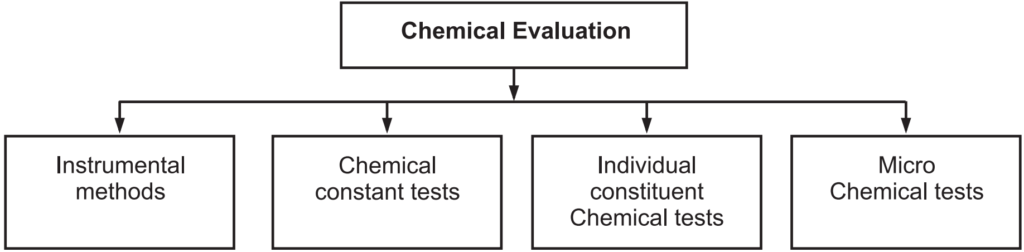
Instrumental methods:
Various instrumental methods like colorimetry, fluorimetry, spectrophotometry, etc. are used for the evaluation.
Colorimetric Method: It is a method of determining the concentration of a chemical element or chemical compound in a solution with the aid of a color reagent. It is applicable to both organic and inorganic compounds and may be used with or without an enzymatic stage. The method is widely used in medical laboratories and for industrial purposes, e.g., the analysis of water samples in connection with industrial water treatment.
Photometric Method: It is the set of methods of quantitative chemical analysis based on the relationship between the concentration of a substance in a solution or gas and the absorption of radiation. In this method, the intensities of the monochromatic components of transmitted radiation are scanned.
Fluorimetric Method: It is an analytical technique for identifying and characterizing minute amounts of a substance by excitation of the substance with a beam of ultraviolet light and detection and measurement of the characteristic wavelength of fluorescent light emitted.
Gravimetric Method: It is the quantitative determination of a substance by the precipitation method of gravimetric analysis involving isolation of an ion in solution by a precipitation reaction, filtering, washing the precipitate, conversion of precipitate to a product of known composition, and finally weighing the precipitate and determining its mass by difference. There are four fundamental types of gravimetric analysis: physical gravimetry, thermogravimetry, precipitative gravimetric analysis, and electrodeposition.
Volumetric Method: It is a quantitative analysis of liquids or solutions by comparing the volumes that react with known volumes of standard reagents, usually by titration. A reagent is prepared as a standard solution, acts as a titrator. A known concentration and volume of titrant react with a solution of analyte or titrated to determine concentration.
Chemical Constant Tests:
Various tests like acid value, iodine value, Saponification value, etc. are used for the evaluation of fixed oils and fats.
Saponification value: It is the number of milligrams of potassium hydroxide required to saponify 1g of fat under the specific conditions. It is a measure of the average molecular weight of all the fatty acids present.
Saponification value = 28.05 (y – x)/w
where, w = Weight of substance in g.
Acid value: It is the number of milligrams of potassium hydroxide (KOH) necessary to neutralize the fatty acids in 1 gram of sample.
Acid value: 5.61 n/w
where, n = mL of 0.1 m Potassium hydroxide solution required. w = Weight of sample in g.
Significance:
- to know the freshness of the oil.
- to know the degree of rancidity of oil.
Iodine value: It is the mass of iodine in grams that is consumed by 100 grams of a chemical substance. Iodine numbers are often used to determine the amount of unsaturation in fatty acids. The higher the iodine number, the more C=C bonds are present in the fat.
Iodine value = 1.269 (y – x)/w where w is the weight of sample in g.
Significance:
- To determine the quality of any unsaturated oils.
- It measures the degree of unsaturation in a fat or oil.
- It provides a degree of rancidity.
Individual Constituent Chemical Tests:
Various chemical tests are carried out for the identification of individual components. For each plant drug, there is some common test for identification of a group of plant secondary metabolites as well as specific tests for identification of individual constituents.
Microchemical Tests:
These tests are carried out on the slide. Example: Eugenol in clove oil is precipitated as potassium euginate when potassium hydroxide is added on a slide containing clove oil. Under microscope clove oil shows needle-shaped crystal of potassium euginate Molish test for detection of sugars, Lieberman Burchard test for detection of steroids, Borntrager test for detection of anthraquinones, ferric chloride test for tannins, Keller- Kiliani test for deoxy sugars, ninhydrin test for the detection of amino acids and proteins, etc.
Biological Evaluation
When the estimation of the potency of a crude drug or its preparation is done by means of its effect on living organisms like bacteria, fungal growth, or animal tissue or entire animal, it is known as a bioassay. This method is generally called for when standardization is not adequately done by chemical or physical means and also for conformity of therapeutic activity of raw material and finished product. In other words, a bioassay is the measure of the sample being tested capable of producing a biological effect as that of the standard preparation. Such activity is represented in units known as an international unit (I.U.).
The specific biological activity contained in each I.U. of the few drugs is mentioned as under:
Digitalis- 1IU is contained in 76 mg of standard preparation
VitA- 1 IU is present in 0.344 micrograms of standard preparation
Biological assay methods are mainly of 3 types
- Toxic
- Symptomatic
- Tissue methods
In toxic and symptomatic techniques, the animals are used, whereas, in the tissue method, the effect of a drug is observed on isolated organs or tissue. Among the drugs that are subjected to bioassay are cardiac glycosides, natural pesticides, and antibiotics.
Make sure you also check our other amazing Article on : Glycosides In Pharmacognosy