
Pharmaceutical manufacturers take several steps to make sure they produce good quality products. Yet, it has been found that several concerns plague the drug development and manufacturing processes. The pioneer in quality, Dr. Joseph M. Juran was the first to develop the concept of Quality by Design (QbD). He proposed that quality must be designed into the product; if this is done, there will not be any of the quality crises that one commonly encounters.
In a report titled, ‘Pharmaceutical Quality for the 21st Century: A Risk-Based Approach,’ the US Food and Drug Administration (FDA) furthered these ideas and provided an initiative to address quality issues. Through collaboration with major pharma companies, the FDA created a set of guidance documents on the concept of Quality by Design (QbD). These were then accepted by the International Conference on Harmonization (ICH) to streamline and regulate the process of drug development and regulatory filing related to drug manufacture.
The main idea behind the QbD concept is that a process must be designed to produce quality products. This becomes possible only when the process and product are both thoroughly understood, the risks involved in its manufacturing are studied carefully, and steps are outlined to mitigate such risks. This QbD approach is significantly different from the traditional, empirical approach that emphasised on testing of quality in the end products.
Table of Contents
Definition of Quality by Design (QbD)
The ICH guideline Q8 (R2) Pharmaceutical Development defines the term Quality by Design (QbD) as “a systematic approach to development that begins with predefined objectives, emphasizes product, process understanding, and process control, based on sound science and quality risk management.”
Overview of Quality by Design (QbD)
From the above definition, it is clear that pharmaceutical QbD is an organized approach to drug development. It starts with pre-determined objectives with an emphasis on understanding the process and product. It focuses on controlling the process and product on the basis of sound science and the concept of quality risk management.
Some of the key objectives of QbD include:
- Achieving meaningful quality specifications for the product on the basis of clinical performance.
- Enhancing process capability, and reducing product defects and variability by improving the process and product design and control.
- To promote root cause analysis and manage any changes of drug product after it has been approved.
- To improve the efficiency of the processes involved in product development and manufacturing.
Thus, in the pharmaceutical QbD approach, first, critical quality characteristics (from the patient’s point of view) need to be identified. These characteristics are then translated into critical quality attributes (CQAs) of the drug product. Next, a relationship is established between manufacturing process variables and CQAs. Successful control of these variables ensures the consistent delivery of a quality drug product with all desired CQAs to the patient.
Elements of QbD
The elements of QbD include:
- Quality Target Product Profile (QTPP) – it identifies the CQAs of the drug product.
- Product design and identifying Critical Material Attributes (CMAs).
- Process design and identifying Critical Process Parameters (CPPs). This includes linking the CMAs and CPPs with CQAs.
- Controls strategy: developing specifications for active pharmaceutical ingredients (APIs), excipients, and final drug product; also controls for every step of the production process.
- Process capabilities and continuous improvement.

Quality Target Product Profile (QTPP):
QTPP is a summary of the quality parameters that must be present in the drug product to ensure the desired quality is achieved. This is the basis on which product design will commence. When formulating the QTPP, the points to be considered include:
- The intended use of the product, its route of administration, desired dosage form, and system used for drug delivery.
- Strength of the dose.
- Container-closure system to be used.
- Release of the therapeutic component and factors that will influence pharmacokinetic parameters (such as dissolution of drug) in the proposed dosage form.
- Quality criteria for the final product – stability, purity, sterility, drug release, etc.
Critical Quality Attributes (CQAs):
After finalizing the QTPP, it is possible to identify the CQAs of the drug product. CQAs are properties of the finished product – physical, chemical, biological or microbiological – that must lie within a certain range, limits, or distribution, in order to ensure that desired quality of the product is attained.
Some examples of quality attributes of drug products include the identity of drug, assay values, content uniformity, drug release profile, degradation products, microbial levels, moisture content, and physical properties such as size, color, shape, and friability. Not all of them may be critical attributes. Whether an attribute is critical or not depends upon the severity of the damage that will be caused if the product falls outside the acceptable range for that particular attribute.
Product Design:
A well-designed product is one that meets patients’ requirements and this can be confirmed through clinical studies. Such a product will maintain its performance throughout its shelf life, and this can be confirmed by stability studies. Thus, product design must be geared towards developing a robust product that delivers the desired QTPP over the entire shelf life of the product.
For good product design, it is important to study the following in detail:
- Physical, chemical, and biological characteristics of the drug (examples: particle size, polymorphism, solubility, melting point, pKa, oxidative stability, partition coefficient, bioavailability, membrane permeability, etc.).
- Type of excipients and their grade, and details of intrinsic excipient variability (common excipients such as binders, diluents, disintegrants, glidants, coloring agents, sweeteners, suspending agents, film coatings, preservatives, flavors, etc.).
- Interactions of drug substances with excipients by carrying out drug-excipient compatibility testing.
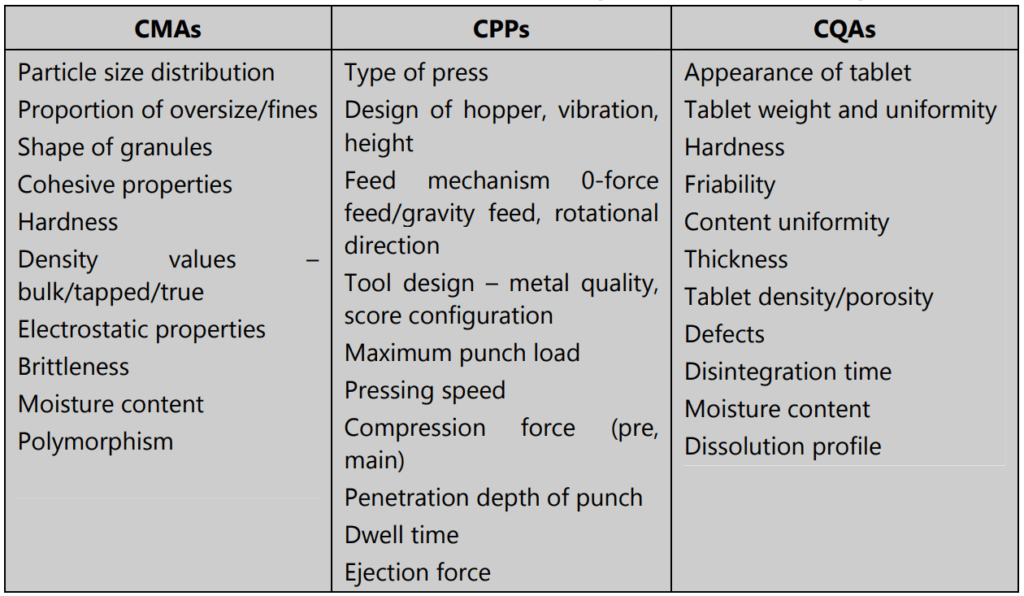
- The critical material attributes (CMAs) of both drug and excipients ensure the development of a robust formulation.
CMA vs CQA

Process Design:
The manufacturing process for a drug product is made up of a set of unit operations run in a particular sequence, to give the final product. The term unit operation refers to any activity where there is a physical or chemical change in the substance. Milling, mixing, granulation, drying, tablet compression, coating, are all examples of unit operations in tablet manufacture.
Processes must be designed in such a way that each unit operation is performed as expected to deliver the necessary product. For this, it is important to:
- Identify the critical causes of variations.
- Manage these variations during the process.
- Predict quality attributes of the product with accuracy and reliability.
Any parameter whose variability can have an adverse impact on a CQA is critical to the process and called a Critical Process Parameter (CPP). All CPPs for a given process must be first identified; then they must be monitored and regulated to make sure that desired quality products are produced.
Process robustness studies must be performed to check if the process can tolerate variability in the input materials and processing parameters and still deliver a product of acceptable quality. These studies will also serve to identify CPPs which have an impact on drug quality.
How to understand processes?
- List all process parameters that may impact the process performance
- Using scientific knowledge and risk assessment, identify the parameters that are potentially high risk
- Establish ranges for these high-risk potential parameters
- Design and carry out experiments to test these parameters
- Obtain experimental data and analyze it using first-principle models to confirm how critical the process parameter is. Connect CPPs and CMAs to CQAs wherever possible
- Develop a control mechanism by defining acceptable ranges for critical parameters and non-critical parameters.
Control Strategy:
The data generated during developmental studies must be used to set up a control strategy. It is common to have controls at three levels as follows:
Level 1: Automated engineering controls are used for real-time monitoring of CQAs of the output materials. The system is designed to monitor the input material attributes and adjust the process parameters automatically so that CQAs consistently meet the predetermined acceptance criteria. Process Analytical Technology (PAT) systems are an example of this type of control.
Level 2: Here, the emphasis is on understanding the process and product, and designing it with control over the pharmaceutical process. This is QbD and it allows the control of variables, and thus, ensures drug product quality.
Level 3: This strategy depends on detailed testing of end-product as seen in conventional pharmaceutical manufacturing. As the sources of variability have not been identified, and there is no study of CMAs and CPPs on the quality of drug products, the likelihood of product problems is high.
In real-life situations, it is best to combine level 1 and level 2 control strategies to arrive at a hybrid approach that involves:
- Controlling attributes of input material based on a study of their impact on product quality and processability.
- Establishing product specifications.
- Controlling unit operations that have the biggest impact on product quality.
- Testing in-process, in real-time instead of relying on end-product testing.
- Setting up a monitoring program to verify control over the process and product.
Process Capability and Continued Improvement:
Process capability is a measure of the level of inherent variability shown by a stable process that is under control when compared with the established acceptance criteria. Variability may be short-term or long-term, and the QbD program must result in the identification and reduction of the variations that impact the quality of the product.
Continuous improvement methods need to be adopted to remove these sources of variability. This includes several activities in different phases such as:
- Defining the problem and set up specific goals
- Measuring key areas of the process and collecting necessary data
- Data analysis to find cause-effect relationships
- Use results of data analysis to optimize the process
- Perform pilot runs to check optimized process capabilities
- Monitor processes to make sure they stay in a state of statistical control
Quality by Design Tools
Quality by Design relies on the use of certain tools. These include prior knowledge, risk assessment, mechanistic models, design of experiments and data analysis, and process analytical technology.
Prior Knowledge
As per ICH guidelines, prior knowledge is the information or knowledge or skills that have been acquired through previous experience of similar processes and published information. This tool can be used at the beginning of the developmental process and maybe regularly updated using data generated during the process. Prior knowledge can be applied as a part of control strategies, in relation to QTPP and CQAs. However, it is important to avoid too much reliance on prior knowledge as it may result in a loss of control over the manufacturing process. It is best to use this tool to confirm data rather than to build data from scratch.
Risk Assessment
As per ICH Q9, quality risk management must be done before development studies to detect the high-risk variables that have an impact on drug product quality. Risk evaluation must be done on the basis of scientific knowledge and is often used to determine critical variables. These variables must then be further investigated through experimentation, so that a control strategy may be established.
Some of the common risk assessment tools used are flowcharts, fault tree analysis, failure mode effects analysis, hazard analysis and critical control points, risk ranking and filtering, etc.
FMEA and HACCP
Failure Mode Effect Analysis (FMEA): Failure mode refers to the defects or errors in material, equipment, design, or process. After establishing these failure modes, the tool evaluates their effects and ranks them in order of priority. This method may also include a study of how critical the consequences of the failures are. Sometimes, Ishikawa diagrams (fishbone/cause-and-effect) are also used.
Hazard Analysis and Critical Control Points (HACCP): Hazards that can cause safety and quality issues are identified (for example, hygiene of personnel, material flow, environmental aspects, process design, manufacturing steps). Preventive measures for each of these are established. Next, critical control points are determined for these hazards, and limits are established. A system is set up to monitor these critical control points, and corrective actions to be taken when these are not in a state of control, are determined. Finally, record-keeping systems are set up to monitor and confirm that the HACCP system itself is working as expected.
Design of Experiments
This tool involves setting up a series of structured tests where changes to the variables of a process are made in a planned manner. Then, the impact of these changes on a chosen output is assessed. This tool is very effective in identifying all the factors that together impact the output responses. The interaction of the variable factors can also be quantified.
Process Analytical Technology (PAT)
The US FDA defines PAT as “A system for designing, analyzing, and controlling manufacturing through timely measurements (i.e., during processing) of critical quality and performance attributes of raw and in-process materials and processes, with the goal of ensuring final product quality.
PAT allows real-time monitoring of CMAs, CPPs, or CQAs to demonstrate that the process is in a state of control. It enables online measurements that are very useful to detect failures, and also allows adjustment of the operational parameters when variations that have a negative impact on product quality are detected.
PAT includes a wide variety of tools to acquire physical, chemical, microbiological, analytical, and mathematical data and risk analysis. By creating an interface of the process with the instrument, and also a feedback loop that can modify processing conditions, PAT helps to control process parameters as well as product quality.
Advantages of QbD:
- Better assurance of product quality due to improved process design and better quality risk management during the manufacturing process.
- Innovation and increased efficiency and reduced potential for errors lead to cost savings.
- Improves regulatory compliance and streamlines change management.
- Real-time testing during the process ensures faster releases as compared to traditional end-testing of finished products.
Challenges to QbD:
- Requires a cultural change in the organizational approach to quality.
- Expensive requires management support.
- Calls for collaboration between departments and there may be resource/workload limitations.
In conclusion, we can understand QbD as a quality system that helps to manage the life cycle of a product. It aims at designing a capable process through better product and process understanding and through this, hopes to reduce the risk of patients taking drug products. The emphasis in QbD is on continuous improvement, building on past experience, using risk management approaches, and documenting knowledge to achieve high-quality drug products that consistently meet their quality specifications.
Make sure you also check our other amazing Article on: International Conference On Harmonization