Cell injury is the common denominator in almost all diseases. It is defined as “an alteration in cell structure or biochemical functioning, resulting from some stress that exceeds the ability of the cell to compensate through normal physiologic adaptive mechanisms”.
Cell injury results when cells are stressed so severely that they are no longer able to adapt or when cells are exposed to inherently damaging agents or suffer from intrinsic abnormalities. Different injurious stimuli affect many metabolic pathways and cellular organelles. Injury may progress through a reversible stage and culminate in cell death.
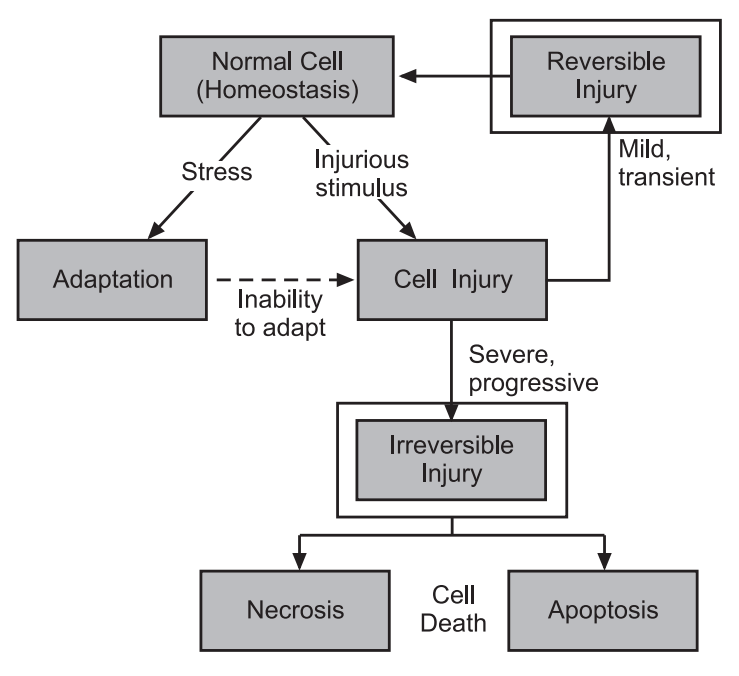
Table of Contents
Types of Cell injury
Reversible cell injury
In early stages or mild forms of injury, the functional and morphologic changes are reversible if the damaging stimulus is removed. At this stage, although there may be significant structural and functional abnormalities, the injury has typically not progressed to severe membrane damage and nuclear dissolution.
Irreversible cell injury (Cell death)
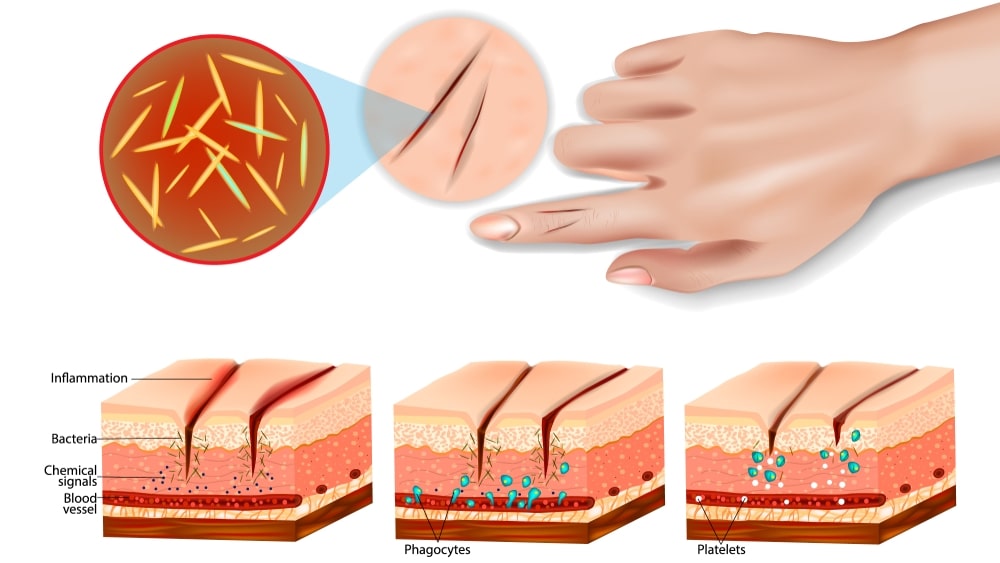
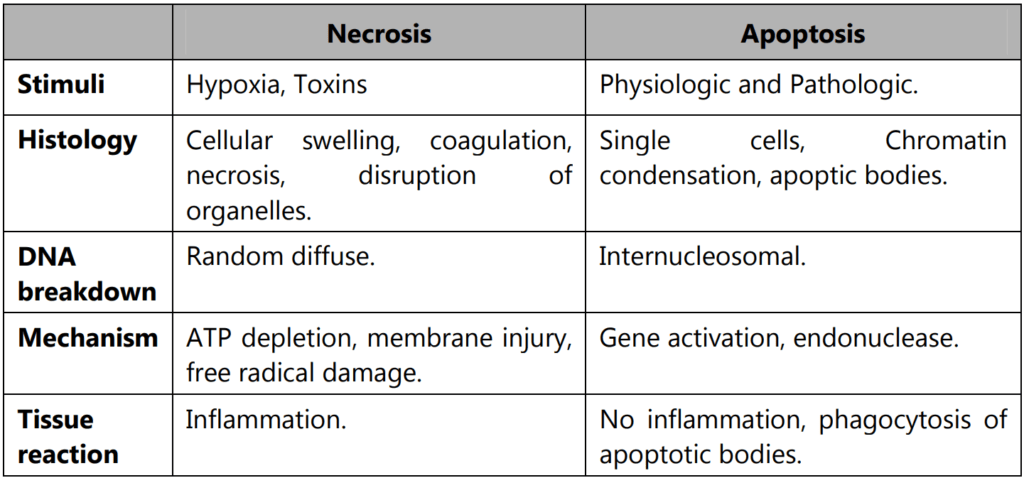
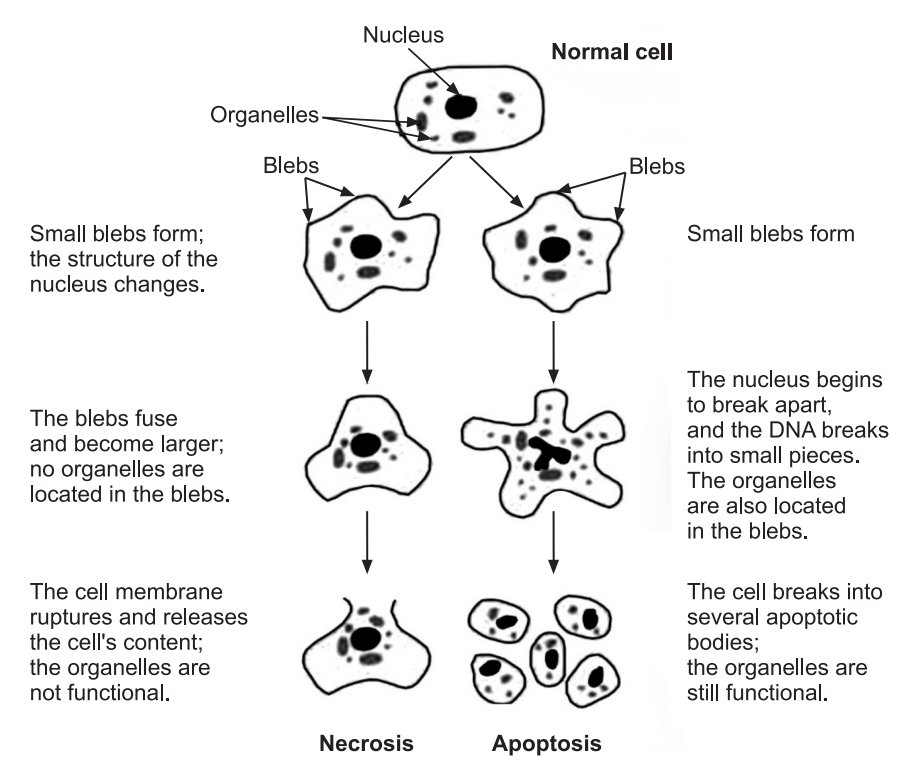
Because of cell death with continuing damage, the injury becomes irreversible, at which time the cell cannot recover and it dies. There are two types of cell death, necrosis, and apoptosis which differ in their morphology, mechanisms, and roles in disease and physiology. When damage to membranes is severe, enzymes leak out of lysosomes, enter the cytoplasm, and digest the cell, resulting in necrosis. Cellular contents also leak out through the damaged plasma membrane and elicit a host reaction (inflammation). Necrosis is the major pathway of cell death in many commonly encountered injuries, such as those resulting from ischemia, exposure to toxins, various infections, and trauma. When a cell is deprived of growth factors or the cell’s DNA or proteins are damaged beyond repair, the cell kills itself by another type of death, called apoptosis, which is characterized by nuclear dissolution without complete loss of membrane integrity. Apoptosis is an active, energy-dependent, tightly regulated type of cell death that is seen in some specific situations. Whereas necrosis is always a pathologic process, apoptosis serves many normal functions and is not necessarily associated with pathologic cell injury.
Necrosis
Necrosis is one of the basic patterns of irreversible cell injury and death. Necrosis has long been considered the “unregulated” pattern of cell injury and death, representing a messy end to a damaged cell that consequently causes a potent inflammatory response.
Apoptosis
This is a pathway of cell death that is induced by a tightly regulated suicide program in which cells are destined to die by activating enzymes capable of degrading the cell’s own nuclear DNA, nuclear and cytoplasmic proteins. Fragments of the apoptotic cells then break off; giving the appearance that is responsible for the name (apoptosis, “falling off”). The plasma membrane of the apoptotic cell remains intact, but the membrane is altered in such a way that the cell and its fragments become avid targets for phagocytes. The dead cell is rapidly cleared before its contents have leaked out, and therefore cell death by this pathway does not elicit an inflammatory reaction in the host. Thus, apoptosis differs from necrosis, which is characterized by loss of membrane integrity, enzymatic digestion of cells, leakage of cellular contents, and frequently a host reaction. However, apoptosis and necrosis sometimes coexist, and apoptosis induced by some pathologic stimuli may progress to necrosis.
Etiology of Cell Injury
Cell injury is a sequence of events that occur if the limits of adaptive capability are exceeded or no adaptive response is possible. This can be due to physical, chemical, infectious, biological, immunological factors, and nutritional cellular abnormalities.
Acquired cause:
Acquired causes of cell injury are further categorized as:
- Oxygen deprivation (Hypoxia)
- Physical agents
- Chemical agents and drugs
- Microbial agents
- Immunologic agents
- Nutritional derangement
- Psychological factors
- Idiopathic agents
Oxygen deprivation: Hypoxia is a deficiency of oxygen, which causes cell injury by reducing aerobic oxidative respiration. Hypoxia is an extremely important and common cause of cell injury and cell death. Causes of hypoxia include reduced blood flow (ischemia), inadequate oxygenation of the blood due to cardiorespiratory failure, and decreased oxygen-carrying capacity of the blood, as in anemia or carbon monoxide poisoning (producing a stable carbon monoxyhemoglobin that blocks oxygen carriage) or after severe blood loss. Depending on the severity of the hypoxic state, cells may adapt, undergo injury, or die. For example, if an artery is narrowed, the tissue supplied by that vessel may initially shrink in size (atrophy), whereas more severe or sudden hypoxia induces injury and cell death.
Physical agents for cell injury: Mechanical trauma (e.g., Road accident), Thermal trauma (e.g., Heat and cold), Electricity, Radiation (e.g., U.V. radiation), Rapid changes in atmospheric pressure.
Chemicals and Drugs: The list of chemicals that may produce cell injury defines compilation. Simple chemicals such as glucose or salt in hypertonic concentrations may cause cell injury directly or by deranging electrolyte balance in cells. Even oxygen at high concentrations is toxic. Trace amounts of poisons, such as arsenic, cyanide, or mercuric salts, may damage a sufficient number of cells within minutes or hours to cause death. Other potentially injurious substances are our daily companions: environmental and air pollutants, insecticides, and herbicides; industrial and occupational hazards, such as carbon monoxide and asbestos; recreational drugs such as alcohol; and the ever-increasing variety of therapeutic drugs.
Microbial agents: Injuries by microbes include infection caused by bacteria, rickettsiae, viruses, fungi, protozoa, and other parasites.
Immunological Agents: The immune system serves an essential function in defense against infectious pathogens, but immune reactions may also cause cell injury. Injurious reactions to endogenous self-antigens are responsible for several autoimmune diseases. Immune reactions to many external agents, such as viruses and environmental substances, are also important causes of cell and tissue injury. Example: Hypersensitivity reactions, anaphylactic reactions, autoimmune diseases.
Nutritional derangement: A deficiency or an excess of nutrients may result in nutritional imbalances. Nutritional deficiency diseases may be due to overall deficiency of nutrients (starvation), protein-calorie (Marasmus, Kwashiorkor), and minerals (Anaemia) or of trace elements. Nutritional excess is a problem of a society that results from obesity, atherosclerosis, heart diseases, and hypertension.
Psychological factors: There is a number of specific biochemical or morphological changes in common acquired mental diseases due to mental stress, strain, anxiety, overwork, and frustration. Problems of drug addiction, alcoholism, and smoking result in various diseases such as liver damage, chronic bronchitis, lung cancer, peptic ulcer, hypertension, ischemic heart diseases, etc.
Idiopathic factor: The causative factor of cell injury is unknown.
Genetic cause:
In western countries, genetic defects constitute about 50% of total mortality in infancy and childhood, while in developing and under-developing countries 95% of infant mortality occurs. Genetic causes are such as, Developmental defects (Errors in morphogenesis), cytogenic defects (chromosomal abnormalities), Single-gene defects (Mendelian syndrome), Storage diseases (Inborn Errors of metabolism), Disorders with multifactorial inhabitation.
Pathogenesis of Cell Injury
Cell damage can be reversible or irreversible. Depending on the extent of injury, the cellular response may be adaptive and homeostasis is maintained. Cell death occurs when the severity of the injury (Stress) exceeds the cell’s ability to repair itself. Cell death is relative to both the length of exposure to a harmful stimulus and the severity of the damage caused. Cell death may occur by severe cell swelling or cell rupture, denaturation and coagulation of cytoplasmic proteins, and breakdown of cell organelles (necrosis) or internally controlled cell death, chromatin condensation, and fragmentation (apoptosis). Now, we have discussed the causes of cell injury and necrosis and their morphologic and functional correlates, we next consider in more detail the molecular basis of cell injury and then illustrate the important principles with a few selected examples of common types of injuries. The biochemical mechanisms linking any given injury with the resulting cellular and tissue manifestations are complex, interconnected, and tightly interwoven with many intracellular metabolic pathways. It is therefore often difficult to pinpoint specific molecular alterations caused by a particular insult.
The cellular response to injurious stimuli depends on the type of injury, its duration, and its severity. Thus, low doses of toxins or a brief duration of ischemia may lead to reversible cell injury, whereas larger toxin doses or longer ischemic intervals may result in irreversible injury and cell death.
The consequences of an injurious stimulus depend on the type, status, adaptability, and genetic makeup of the injured cell. The same injury has vastly different outcomes depending on the cell type; thus, striated skeletal muscle in the leg accommodates complete ischemia for 2 to 3 hours without irreversible injury, whereas cardiac muscle dies after only 20 to 30 minutes. The nutritional (or hormonal) status can also be important; clearly, a glycogen-replete hepatocyte will tolerate ischemia much better than one that has just burned its last glucose molecule. Genetically determined diversity in metabolic pathways can also be important. For instance, when exposed to the same dose of a toxin, individuals who inherit variants in genes encoding cytochrome P-450 may catabolize the toxin at different rates, leading to different outcomes. Much effort is now directed toward understanding the role of genetic polymorphisms in responses to drugs and toxins and in disease susceptibility.
Cell injury results from functional and biochemical abnormalities in one or more of several essential cellular components. The most important targets of injurious stimuli are:
- Mitochondria, the sites of ATP generation
- Cell membranes, on which the ionic and osmotic homeostasis of the cell and its organelles depends;
- Protein synthesis;
- The cytoskeleton; and
- The genetic apparatus of the cell.
Mechanism of Reversible Cell Injury
As the name implies, this occurs if extreme stress persists and the cell is unable to adapt to overcome the stress. Reversible cell injury results in cellular and morphological changes that can still be reversed if the stress is eventually removed. There are three mechanisms by which reversible cell injury may occur.
- Depleted resources of ATP in the cell owing to decreased levels of Oxidative Phosphorylation.
- Hydropic cellular swelling is a phenomenon caused by changes in ion concentrations and water influx.
- Organelles within the cell show minute alterations.
ATP depletion:
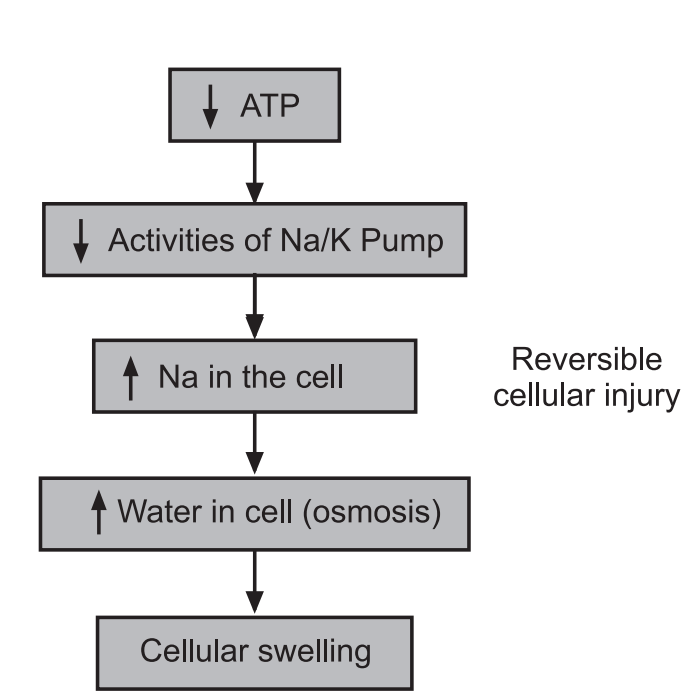
ATP depletion and decreased ATP synthesis are frequently associated with both hypoxic and chemical (toxic) injury. High-energy phosphate in the form of ATP is required for many synthetic and degradative processes within the cell. These include membrane transport, protein synthesis, lipogenesis, and the diacylation deacylation reactions necessary for phospholipid turnover. Depletion of ATP to 5% to 10% of normal levels has widespread effects on many critical cellular systems. The activity of the plasma membrane energy-dependent sodium pump is reduced. Failure of this active transport system, due to diminished ATP concentration and enhanced ATPase activity, causes sodium to accumulate intracellularly and potassium to diffuse out of the cell. The net gain of solute is accompanied by isosmotic gain of water, causing cell swelling, dilation of the endoplasmic reticulum, and cellular energy metabolism is altered. If the supply of oxygen to cells is reduced, as in ischemia, oxidative phosphorylation ceases and cells rely on glycolysis for energy production.
This switch to anaerobic metabolism is controlled by energy pathway metabolites acting on glycolytic enzymes. The decrease in cellular ATP and the associated increase in adenosine monophosphate stimulate phosphofructokinase and phosphorylase activities. These result in an increased rate of anaerobic glycolysis designed to maintain the cell’s energy sources by generating ATP through the metabolism of glucose derived from glycogen. As a consequence, glycogen stores are rapidly depleted.
Glycolysis results in the accumulation of lactic acid and inorganic phosphates from the hydrolysis of phosphate esters. This reduces the intracellular pH, resulting in decreased activity of many cellular enzymes.
Damage to Mitochondria: Mitochondria are the cell’s suppliers of life-sustaining energy in the form of ATP, but they are also critical players in cell injury and death. Mitochondria can be damaged by the increase of cytosolic Ca2+, reactive oxygen species, and oxygen deprivation, and so they are sensitive to virtually all types of injurious stimuli, including hypoxia and toxins.
There are two major consequences of mitochondrial damage:
- Mitochondrial damage often results in the formation of a high-conductance channel in the mitochondrial membrane, called the mitochondrial permeability Mitochondrial damage often results in the formation of a high-conductance channel in the mitochondrial membrane, called the mitochondrial permeability transition pores. The opening of this channel leads to the loss of mitochondrial membrane potential and pH changes, resulting in failure of oxidative phosphorylation and progressive depletion of ATP, culminating in necrosis of the cell.
- The mitochondria also contain several proteins that are capable of activating apoptotic pathways, including cytochrome C (the major protein involved in electron transport). Increased permeability of the mitochondrial membrane may result in leakage of these proteins into the cytosol and death by apoptosis. Thus, cytochrome C plays a key dual role in cell survival and death. In its normal location inside mitochondria, it is essential for energy generation and the life of the cell, but when mitochondria are damaged so severely that cytochrome C leaks out, it signals cells to die.
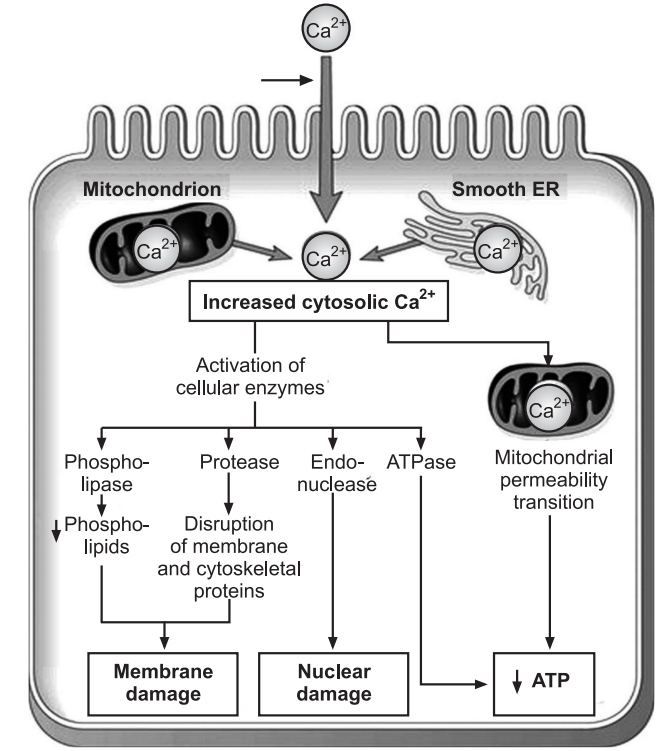
The influx of Calcium:
Failure of the Ca2+ pump leads to an influx of Ca2+, with damaging effects on numerous cellular components. With prolonged or worsening depletion of ATP, structural disruption of the protein synthetic apparatus occurs, manifested as detachment of ribosomes from the rough endoplasmic reticulum and dissociation of polysomes into monosomes, with a consequent reduction in protein synthesis. Ultimately, there is irreversible damage to mitochondrial and lysosomal membranes, and the cell undergoes necrosis.
In cells deprived of oxygen or glucose, proteins may become misfolded, and misfolded proteins trigger a cellular reaction called the unfolded protein response that may lead to cell injury and even death. A protein misfolding is also seen in cells exposed to stress, such as heat, and when proteins are damaged by enzymes and free radicals.
- Defects in membrane permeability: Early loss of selective membrane permeability leading ultimately to overt membrane damage is a consistent feature of most forms of cell injury. The plasma membrane can be damaged by ischemia, various microbial toxins, lytic complement components, and a variety of physical and chemical agents. Several biochemical mechanisms may contribute to membrane damage.
- Decreased phospholipid synthesis: The production of phospholipids in cells may be reduced whenever there is a fall in ATP levels, leading to decreased energy-dependent enzymatic activities. The reduced phospholipid synthesis may affect all cellular membranes including the mitochondria themselves, thus exacerbating the loss of ATP.
- Increased phospholipid breakdown: Severe cell injury is associated with increased degradation of membrane phospholipids, probably due to activation of endogenous phospholipases by increased levels of cytosolic Ca2+ .
- ROS: Oxygen-free radicals cause injury to cell membranes by lipid peroxidation, discussed earlier.
- Cytoskeletal abnormalities: Cytoskeletal filaments serve as anchors connecting the plasma membrane to the cell interior. Activation of proteases by increased cytosolic Ca2+ may cause damage to elements of the cytoskeleton.
- Lipid breakdown products: These include unesterified free fatty acids, acylcarnitine, and lysophospholipids, catabolic products that are known to accumulate in injured cells as a result of phospholipid degradation. They have a detergent effect on membranes. They also either insert into the lipid bilayer of the membrane or exchange with membrane phospholipids, potentially causing changes in permeability and electrophysiologic alterations. The most important sites of membrane damage during cell injury are the mitochondrial membrane, the plasma membrane, and membranes of lysosomes.
- Mitochondrial membrane damage: As discussed above, damage to mitochondrial membranes results in decreased production of ATP, culminating in necrosis, and release of proteins that trigger apoptotic death.
- Plasma membrane damage: Plasma membrane damage leads to loss of osmotic balance and influx of fluids and ions, as well as loss of cellular contents. The cells may also leak metabolites that are vital for the reconstitution of ATP, thus further depleting energy stores.
- Injury to lysosomal membranes: Injury to lysosomal membranes results in leakage of their enzymes into the cytoplasm and activation of the acid hydrolases in the acidic intracellular pH of the injured (e.g., ischemic) cell. Lysosomes contain RNAases, DNAases, Proteases, Glucosidases, and other enzymes. Activation of these enzymes leads to enzymatic digestion of cell components, and the cells die by necrosis.
- Damage to DNA and proteins: Cells have mechanisms that repair damage to DNA, but if this damage is too severe to be corrected (e.g., after radiation injury or oxidative stress), the cell initiates its suicide program and dies by apoptosis. A similar reaction is triggered by improperly folded proteins, which may be the result of inherited mutations or external triggers such as free radicals. Since these mechanisms of cell injury typically cause apoptosis.
- Reduced protein synthesis: As a result of continued hypoxia, membranes of the endoplasmic reticulum and Golgi apparatus swell up. Ribosomes are detached from the granular endoplasmic reticulum and polysomes are degraded to monosomes, thus dispersing ribosomes in the cytoplasm and inactivating their function. Similarly, reduced protein synthesis occurs in the Golgi apparatus. Up to this point, withdrawal of acute stress that resulted in reversible cell injury can restore the cell to a normal state.
Mechanism of Irreversible Cell Injury
The long-term decrease in oxygenated blood supply results in irreversible damage to the cellular structure and functions. The sequence of the reversible cell injury continues and reaches into irreversible cell damage (cell death). In irreversible cell injury, the inability of the cell to reverse mitochondrial and plasma membrane dysfunction on reperfusion or reoxygenation. Besides this, there is a further reduction in ATP continued depletion to proteins, reduced intracellular pH, and leakage of lysosomal enzymes into the cytoplasm. These biochemical changes alter the normal functions of the cell which are described as follows:
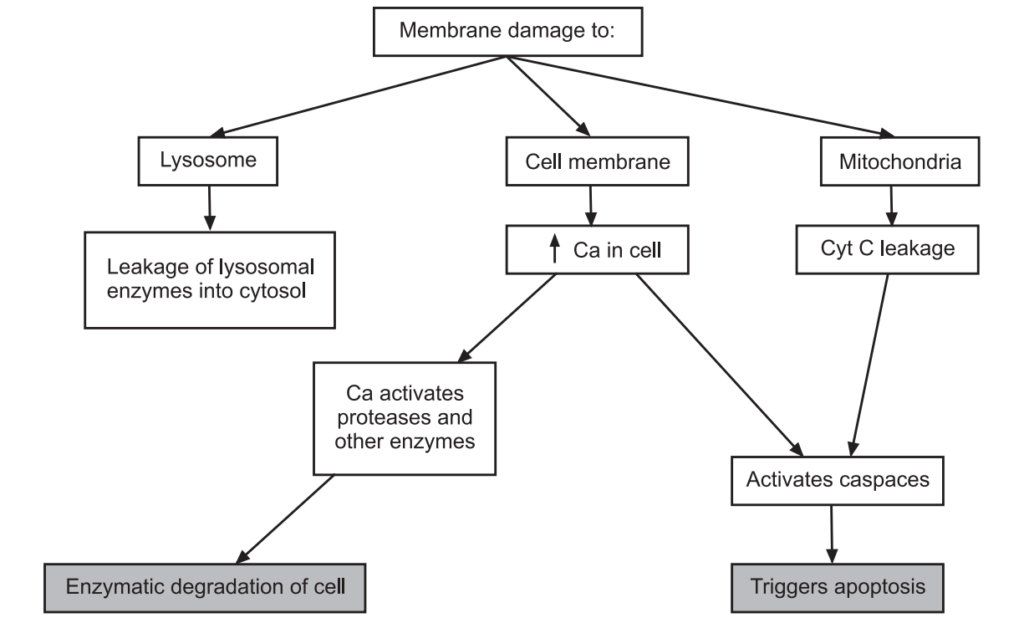
- Mitochondrial damage: As a result of the continued decrease in oxygenated blood supply, irreversible cell damage occurs and on reperfusion with injured cells, excess intracellular calcium collects in the mitochondria disabling its function. Morphologically, mitochondrial changes are vacuoles in the mitochondria and deposition of amorphous calcium salts in the mitochondrial matrix.
- Membrane damage: Damage to plasma membrane losing its normal function is the most important event in irreversible cell injury. Due to damage to the plasma membrane, a cytosolic influx of calcium in the cell increases. Calcium activates endogenous phospholipase. Activated phospholipase degrade membrane phospholipids progressively which are the main constituents of the lipid bilayer membrane. Besides these, activation of lytic enzymes ATPase which causes further depletion of ATP leads to a decrease in the synthesis of new phospholipid for replacement.
- Cytoskeletal damage: Activated intracellular protease or by the physical effect of cell swelling, damages of the cytoskeleton may lead to irreversible cell membrane injury.
- Nuclear damage: The nucleoproteins are damaged by the activated lysosomal enzymes such as proteases and endonucleases. Irreversible damage to the nucleus can be in three forms.
- Pyknosis: Condensation and clumping of a nucleus which becomes dark basophilic.
- Karyorrhexis: Nuclear fragmentation into small bits dispersed in the cytoplasm.
- Karolysis: Dissolution of the nucleus.
5. Lysosomal damage, cell death, and phagocytosis: The lysosomal membranes are damaged and result in the escape of lysosomal hydrolytic enzymes. These enzymes are activated due to the lack of oxygen in the cell and acidic pH. These hydrolytic enzymes include Hydrolase, protease, glycosidase phosphatase, lipase, amylase, RNAase, and DNAase which on activation bring about enzymatic digestion of cellular components and hence cell death. The dead cell is eventually replaced by masses of phospholipids called myelin figures which are either phagocytosed by macrophages or there may be the formation of calcium soaps. Liberated enzymes just mentioned leak across the abnormally permeable cell membrane into the serum, the estimation of which may be used as clinical parameters of cell death. In myocardial infarction, estimation of SGPT, LDH, CKMB, and cardiac troponins useful guides for the death of heart muscle.
Mechanism of Hypoxia-Induced Cell Injury
Hypoxia is caused by inadequate oxygenation to the cell because of the lack of blood supply to a tissue due to thrombosis. Hemorrhage can cause hypoxia by interrupting the blood supply or blood is not getting oxygenated properly, as it occurs in cardiorespiratory failure, and the oxygen-carrying capacity of blood is diminished in carbon monoxide poisoning hypoxia will occur.
The first point of attack of hypoxia is on the cell’s aerobic respiration, in other words, oxidative phosphorylation. Lack of ATP generation leads to an inability of the cell to maintain its ion-transport systems and the cell begins to swell. If the hypoxia continues, extensive damage to the cell membrane and cell death will ensue.
Free Radical Induced Injury
Most agents acting this way cause cell damage by affecting directly cell membranes and triggering a lethal sequence of events. Free radicals are chemical species that have a single unpaired electron in outer orbit, they initiate an autocatalytic reaction which mainly occurs in reperfusion of the ischemic cell. Activated oxygen radicals are now known to be the common mechanism to cells in injury in many conditions, i.e. aging, chemical and radiation injury, bacterial infections, inflammation, tumor necrosis, etc. Free radicals like superoxide radicals, hydroxyl ions, and peroxide ions are very destructive to cells which cause lipid peroxidation, oxidation of protein, DNA damage, cytoskeleton damage, etc. They are initiated within cells by enzymatic reactions and non-enzymatic systems. The system has a series of protective mechanisms to protect the cells from these free radicals like antioxidant enzymes such as catalase, glutathione peroxidase, and superoxide dismutase. Vitamin E and selenium also help for protection from free radical-induced cellular damage.
The deficiency of all these protective mechanisms may lead to free radical reactive cellular damage, especially in muscle.
Different causes for initiation of free radical:
- Ionizing Radiation: Exposure to Ionizing Radiation causes the generation of a variety of free radical species. This occurs following the radiation-induced splitting of molecules which often generates free radical products.
- Enzymatic metabolism of chemicals or drugs. For e.g, carbon tetrachloride can generate [CCl3]* which causes autooxidation of the polyenic fatty acid present within membrane phospholipids.
- Cellular Respiration: Regulated transfer of free radicals is the basis of the Electron Transport Chain that powers Cellular Respiration. Although the free radicals generated during electron transport are tightly controlled, a small amount can escape and cause damage. Escape of free radicals is substantially enhanced when mitochondria are injured which occurs frequently following metabolic cell injury.
- Chemical Cell Injury: Metabolism of several exogenous chemicals can result in the generation of free radicals. Some metals accept or donate electrons (e– ). For e.g. Cu and Fe (Fenton reaction). Nitric oxide (NO) can act as a free radical and convert into highly reactive peroxynitrite anion (ONOO– ) as well as NO2* and NO– 3. Normally, NO can be produced by endothelial, neurons, macrophages, etc.
The redox reactions occur during normal metabolism. For e.g, in respiration, molecular oxygen is reduced to water by accepting 4 electrons. During this process, a small number of toxic intermediates are formed. The free radical reaction can be studied as follows:
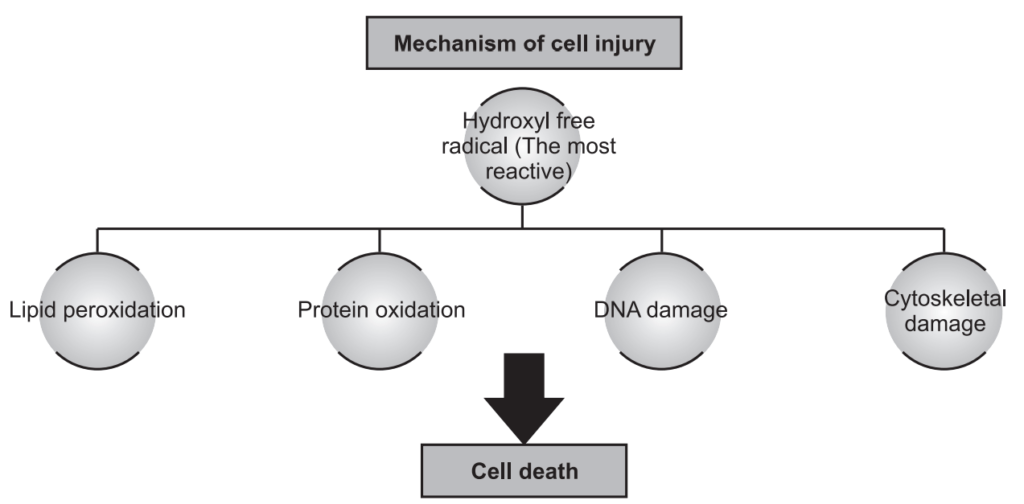
Lipid Peroxidation: Polyunsaturated fatty acid of the membrane is attacked repeatedly by free radicals to form highly destructive polyunsaturated fatty acid (PUFA) radicals like lipid hydroperoxy radicals and lipid hydroperoxides. This is termed as lipid peroxidation. These lipids are widely spread to other parts of the membrane that is lipid peroxidation takes place at the adjoining part of the membrane causing damage to the entire cell membrane.
Oxidation of protein: Free radical causes cleavage by oxidation of protein macromolecules of the cell causing cross-linkage in the amino acid sequences of protein and fragmentation of polypeptides.
Effect on DNA damage: Free radical breaks DNA fragments into a single strand, so there will be the formation of DNA that is defective. Replication of this DNA is not possible and thereby cell death may occur.
Cytoskeleton Damage: Free radicals interfere with mitochondrial aerobic phosphorylation and decrease the synthesis of ATP leading to cytoskeleton damage. There are certain anti-oxidants present endogenously to fight against these oxidative free radicals like Vitamin-E, sulphydryl-containing substances like cystine, SOD, catalase, GTH & serum proteins.
Make sure you also check our other amazing Article on: Formation and Role of ATP